Workflows for Discovering Copy Number Variations Using NGS Data.
Published:
Copy number variations (CNVs) are genomic alterations that involve the gain or loss of specific regions of DNA. These variations can be inherited from parents or can arise de novo, and they play a significant role in neuropsychiatric disorders and cancers.
Traditionally, CNVs have been identified using array-based methods, but with recent advancements in genomic technologies, whole genome sequencing (WGS) approaches are gaining popularity. While existing Next-generation sequencing (NGS)-based methods for CNV detection at the individual sample level often struggle with low detection accuracy, joint modeling approaches that leverage Mendelian transmission within parent-offspring trios have significantly improved accuracy. For those interested, NGS data analysis pipelines for CNV discovery are available on Bitbucket.
| BWA MEM |
| CNAVnator |
| Pindel |
| SVseq2 |
| TrioCNV |
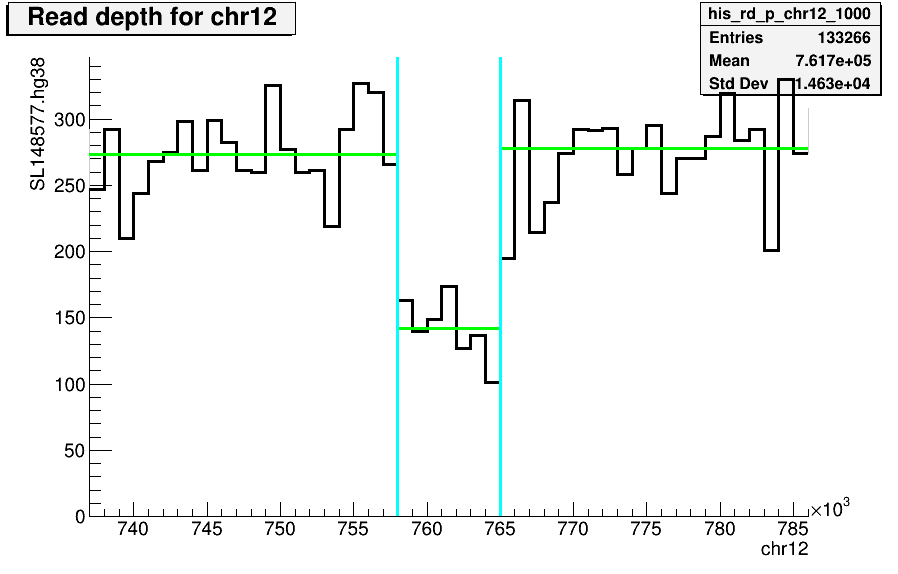
The figure below shows the distribution of average read depth (RD) signal differences between neighboring segments on human chromosome 12 (Chr12:758001-765000).